ISSN Number
ISSN 2771-019X-

-

Impact Factor
1.2*
ISSN Number
ISSN 2771-019X
Impact Factor
1.2*1Hematology and Stem Cell Transplant Unit, Vito Fazzi Hospital, Lecce, Italy.
2Specialist Hematology laboratory, Vito Fazzi Hospital, Lecce, Italy.
Hematology and Stem Cell Transplant Unit, Vito Fazzi Hospital, Lecce, Italy.
Email: miviforina@tiscali.it
Received : Feb 16, 2024,
Accepted : Mar 14, 2024
Published : Mar 21, 2024,
Archived : www.jclinmedcasereports.com
The PICALM-MLLT10 fusion gene, generated by t (10;11) (p12-13; q14-21) translocation, is a rare event, described in various hematologic malignancies. In 2022 ELN classification, PICALM-MLLT10 was identified as acute myeloid leukemia (AML) with other rare recurring translocations. Data on clinical characteristics of PICALM–MLLT10 AML patients are scarce and outcome remains very poor in most patients. We report a case of PICALM-MLLT10 AML with CD7, CD56 co-expression and extramedullary disease.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Dargenio M (2024).
The PICALM-MLLT10 fusion gene, generated by t (10;11) (p12-13; q14-21) translocation, is a rare but recurrent event, observed in patients with Acute Undifferentiated Leukemia (AUL), Acute Lymphoblastic Leukemia (ALL) and AML [1-3]. The literature consists largely of case reports and series of pediatric patients [4]. Rare occurrence, mixed clinical presentation and phenotype with aberrant expression of CD markers associated to immature morphology make the diagnosis challenging. Whereas the 5th edition of the WHO Classification mentioned PICALM-MLLT10 fusions in Mixed Phenotype Acute Leukemia (MPAL) [5], in the International Consensus Classification 2022 PICALM-MLLT10 is not even recognized as a provisional entity [6], although the ELN 2022 classification identifies PICALM-MLLT10 as AML with other rare recurring translocations [7]. Therefore, there is no awareness whether PICALM-MLLT10 corresponds to AML or ALL or acute leukemias of ambiguous lineage (ALAL) and there is no consensus about treatment [5,11]. PICALM–MLLT10 AML were characterized by more frequent extramedullary diseases [2,8,12], CD7 expression and higher platelet counts [8,10]. Treatments were heterogeneuos and driven by their immunophenotype with an outcome remaining very poor in the majority of patients [2,8,12]. Long term responses were achieved in a subset of patients after allogeneic stem-cell transplantation (Allo-HSCT) but also after high-dose cytarabine [8]. We report a case of PICALM-MLLT10 AML with CD7 and concomitant CD56 expression and extramedullary disease.
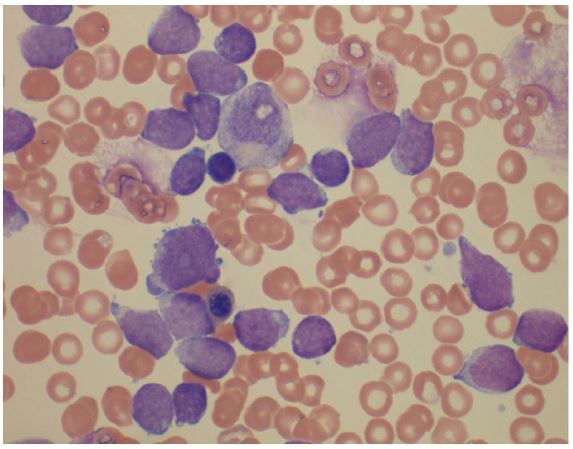
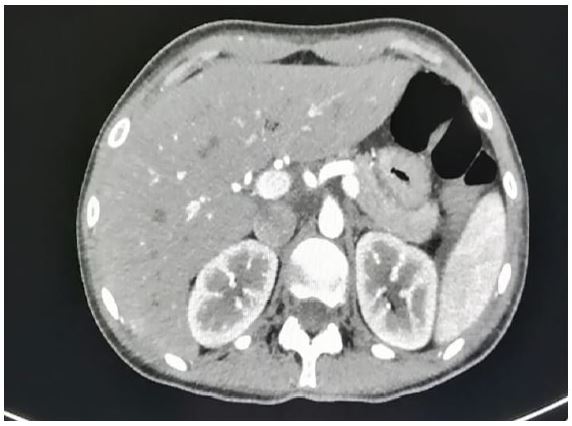
In June 2023, a 26 year old woman was admitted at our hospital with suspicion of Lymphoma. She reported recent EBV infection treated with steroid therapy, weight loss and fever up to 38 degrees centigrade. Physical examination showed bilateral axillary lymphadenopathies and hepatomegaly. HIV serology, PCR testing for CMV and EBV were negative. Blood tests showed a normal lactate dehydrogenase value, a normal hemoglobin, a mild thrombocytopenia (8 5x 10 9/L), white blood cell count of 7. 5 x 109/L with an absolute lymphocytosis of 6.0 x 109/L. Agranular blast cells were found in the peripheral (70%) and bone marrow (90%) smears (Fig.1). Immunophenotypic analysis of both peripheral blood and bone marrow blast cells was positive for CD33, CD34, MPO, HLA-DR and showed a weak positivity for CD5 and aberrant expression of CD7 and CD56, while they were negative for CD117, TdT, CD11c, CD64, CD13, CD68, CD10, CD25, CD20, CD79, CD3, CD4 and CD8. The FISH analysis displayed the t (10;11) (p12-13; q14-21)/PICALM-MLLT10 in 15% of cells analyzed, confirmed by PCR analysis. Nucleophosphomin (NMP1), FMS-like tyrosine kinase 3 (FLT3), RUNX-1 and Core Binding Factor (CBF genes) were negative. Therefore, according to ELN 2022, a diagnosis of AML with other rare recurring translocation was made. A computed tomography scan (CT scan) highlighted multilple lymphadenopathies confluent in a package at the hepatic hilium, at the caval interport, aortic intercavity, left para-aortic and bilateral common iliac (Fig.2), while the cerebrospinal fluid analysis resulted constantly negative for leukemia.The induction therapy consisting of FLAG-IDA regimen was complicated by Pseudomonas Aeruginosa pneumonia resolved after treatment with broad-spectrum antibiotics. At the end of induction therapy, the patient was in complete morphological remission with MRD positivity which became negative after the first consolidation course confirmed on day +60 post-allo-transplant.
The PICALM-MLLT10 rearrangement (alias CALMeAF10) resulting by t (10;11) (p12-13; q14-21) is thought to misdirect DOT1L to the promoters of HOXA genes, leading to hypermethylation of H3K79. This hypermethylation causes constitutive activation of HOXA activity and prevents cell maturation [3,13]. The PICALM-MLLT10 fusion gene has been reported in multilineage blood disease, including AML, ALL, predominantly T-cell, AUL and MPAL, included in Acute Leukemias of Ambiguous Lineage (ALAL) [ 8,11,14]. The 2022 WHO classification includes PICALM-MLLT10 fusions in MPAL [5] and 2022 ELN classification and identifies PICALM-MLLT10 as AML with other rare recurring translocations [7]. Furthermore, there is no separate classification for PICALM MLLT10 in the new ICC classification [6]. Therefore, there is no awareness whether PICALM-MLLT10 corresponds to AML or ALL or ALAL. Unfortunately, sometimes immature morphology, mixed immunophenotype and/or with aberrant expressions and clinical presentation (extramedullary involvement) might confuse and delay the diagnosis [8,13]. Notably in our case, cytogenetic analysis was not indicative due to the absence of metaphases, while FISH and PCR analysis showed t [10,11] (p13; q21)/ PICALM-MLLT10 fusion gene, making for diagnosis more relevant the morphologic, phenotypic and clinical features. The real incidence of PICALM–MLLT10 AML has not been well established in the literature reporting most data concerning pediatric patients [4,8,15]. In the French pediatric ELAM02 protocol, five out of 289 (1.7%) patients had this translocation PICALM–MLLT10 AML in adults might represent 0.3–2% of all AMLs [8]. Compared to negative patient PICALM-MLLT10 positive patient showed younger age, frequent extramedullary involvement, CD7 expression and higher platelets counts in a series of 18 PICALM-MLLT10 AML patients [8]. All of these clinical features were present in our case. Although it is not rare for patients with AML to show aberrant expression of CD 7 or CD 56, it is less common to aberrant co-expression of both markers [10]. Several published experiences suggest the poor prognostic significance of CD7 and /or CD56 expression in patients of AML [16,17] not confirmed in others [18]. It has been reported that the presence of CD56 marker is associated with a higher incidence of CNS disease in patients with AML [19,20], myeloid/ NK AL [21], and ALL [22], while so far there is no evidence of a correlation between CD 56 and PICALM fusion gene [8,16].However, no consensus exists regarding appropriate treatment in PICALM-MLLT10 and often the treatment choice is immunophenotype driven [8,12,15].Children with PICALM-MLLT10 AML have inferior 5-year EFS and OS compared to their T-ALL counterparts [15]. In the French cohort of 18 PICALM-MLLT10 patients treated with AML-like treatments including “3 + 7” induction therapy, the response rate was rather high, and did not differ from that of PICALM-MLLT10 negative AMLs. Relapse rate remained of concern although long term responses can be achieved after Allo-HSCT or high-dose cytarabine [8]. CNS involvement in PICALM Acute Leukemia has been observed at diagnosis as well as at relapse time [4,8,10,15] and justifies careful study of CNS and its prophylaxis regardless of CD 56 expression. In a recent small study, Sun et al showed that a combined therapy including venetoclax might further improve long-term survival of patients undergoing a HSCT in CR even if MRD positive [9]. We treated our patient with the FLAG-Ida regimen, an AML-like regimen, which however contains drugs active on both the lymphoid and myeloid lines. During induction therapy the patient received central nervous system prophylaxis with methotrexate and aracitin and after CR she underwent an allogeneic transplant from an HLA-matched family donor. The FLAG-Ida regimen was also chosen because some studies had demonstrated a higher CR rate and a shorter time to reach it compared to the 3+7 regimen, for the purpose of perfoming the transplant in the shortest time in order to exploit the GVHL effect [23].Currently, 4 months after the transplant, the patient is in CR and a close monitoring is needed due to high post-transplant relapse rate described in these patients [4,8,10,15].The achievement of CR with an AML-like regimen suggests the possible myeloid origin of these leukemias, although recent studies incorporating optical genome mapping (OGM) found PICALM gene also in T-ALL [24].FLAG-Ida regimen may represent an effective and well-tolerated treatment, especially as a bridge to Allo-HSCT. In the near future efforts should be done to exactly clarify the lineage of these rare leukemias by extensive cytogenetic and molecular studies in order to offer the best possible treatment and improve the still dismal outcome of these patients.
Acknowledgments: Published with written consent of the patient.
Conflict of interest: None declared.
Author contributions md and nd: wrote the manuscript. All authors were involved in the care of the patient. All authors read and approved the final manuscript.
Ethical statement: Patient's written informed consent to publication was obtained.
